
1 材料与方法
1.1 样本采集
1.2 种类鉴定和分组
1.3 DNA提取
1.4 文库构建和测序
1.5 数据分析
2 结果
2.1 宏基因组测序质控结果
表1 辽宁省丹东市凤城市采集的耶氏厉螨宏基因组测序数据质量统计Tab. 1 Quality statistics of metagenomic sequencing data of Laelaps jettmari collected from Fengcheng City, Dandong, Liaoning Province |
| 样本编号 | 革螨数量(只) | 原始数据总碱基数(G) | 有效数据总碱基数(G) | Q20(%) | Q30(%) | GC(%) | 有效数据比率(%) |
| G1-1 | 2 | 15.33 | 5.81 | 97.75 | 94.87 | 45.40 | 37.92 |
| G1-2 | 2 | 13.69 | 5.77 | 97.72 | 94.72 | 45.44 | 42.15 |
| G1-3 | 6 | 13.27 | 3.79 | 97.26 | 93.86 | 46.39 | 28.57 |
| G2-1 | 23 | 11.15 | 2.38 | 96.52 | 92.31 | 45.76 | 21.40 |
| G2-2 | 23 | 8.94 | 4.50 | 97.91 | 95.18 | 45.32 | 5.37 |
| G2-3 | 23 | 9.58 | 3.25 | 97.38 | 94.15 | 45.47 | 33.96 |
| G3-1 | 2 | 14.89 | 3.58 | 97.51 | 94.33 | 45.59 | 24.40 |
| G3-2 | 6 | 11.98 | 3.57 | 97.44 | 94.19 | 45.60 | 29.80 |
| G3-3 | 4 | 12.99 | 3.80 | 97.47 | 94.32 | 45.68 | 29.31 |
注:Q20表示质量 > 20的碱基占总数据量的比例;Q30表示质量 > 30的碱基占总数据量的比例;GC表示碱基G和C数量之和占总碱基数量的百分比;有效数据比率表示有效测序序列数占原始测序序列数的比例。 |
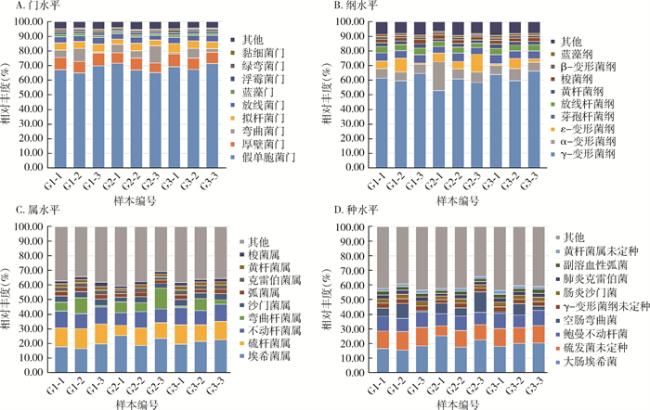
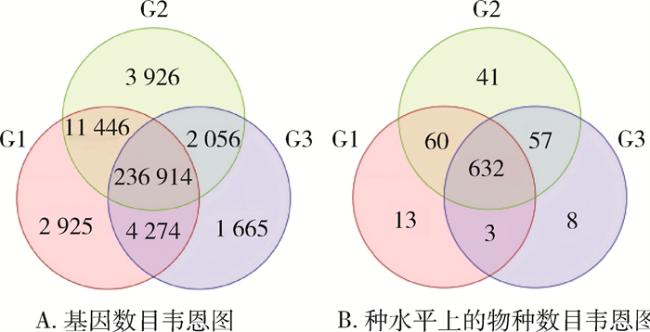
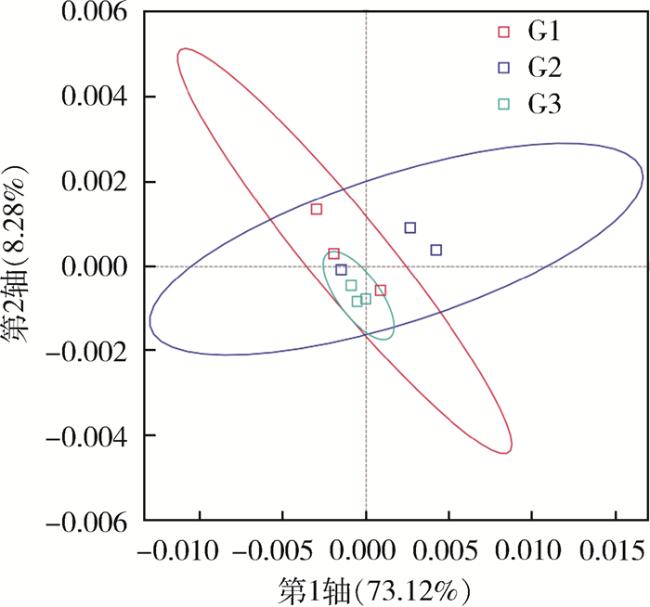
2.2 携带细菌的物种复杂度分析
表2 辽宁省丹东市凤城市采集的耶氏厉螨携带微生物的α多样性Tab. 2 Alpha diversity of microorganisms carried by Laelaps jettmari collected from Fengcheng City, Dandong, Liaoning Province |
| 样本编号 | ACE指数 | Chao1指数 | 香农-威纳指数 | 辛普森指数 | 覆盖度 |
| G1-1 | 1 358.737 | 1 367.607 | 7.672 | 0.979 | 0.999 |
| G1-2 | 1 257.652 | 1 265.027 | 7.506 | 0.977 | 1.000 |
| G1-3 | 1 236.403 | 1 249.270 | 7.677 | 0.978 | 0.999 |
| G2-1 | 1 393.139 | 1 391.598 | 7.518 | 0.969 | 0.999 |
| G2-2 | 1 308.144 | 1 329.553 | 7.694 | 0.979 | 0.999 |
| G2-3 | 1 296.259 | 1 303.412 | 7.198 | 0.966 | 0.999 |
| G3-1 | 1 239.179 | 1 244.766 | 7.710 | 0.979 | 0.999 |
| G3-2 | 1 324.508 | 1 338.140 | 7.744 | 0.979 | 0.999 |
| G3-3 | 1 256.591 | 1 266.000 | 7.606 | 0.976 | 1.000 |
注:ACE指数指abundance-based coverage estimator。 |
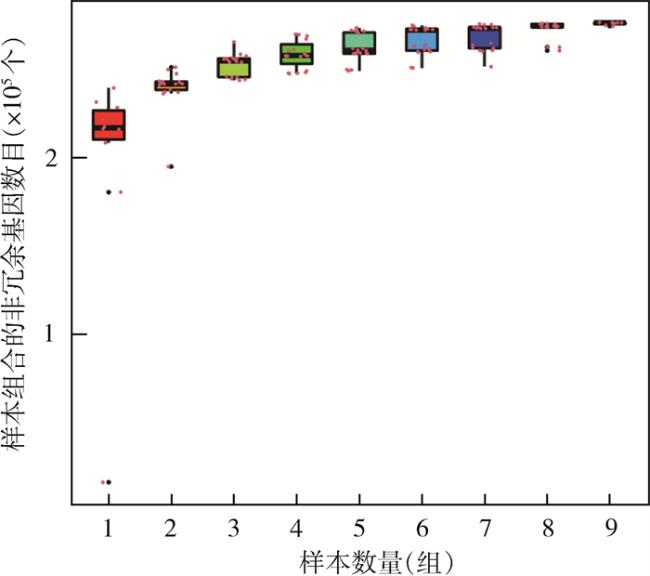
2.3 细菌群落结构及组成分析
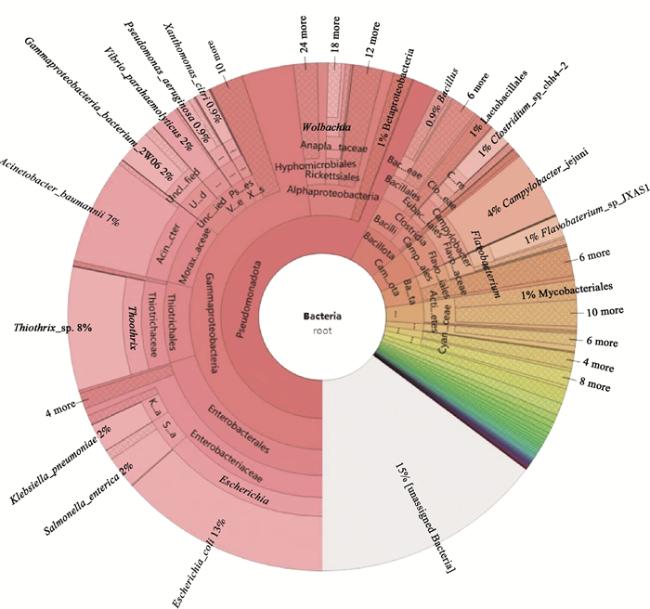
图2 辽宁省丹东市凤城市采集的耶氏厉螨宏基因组测序细菌Krona物种注释可视化结果注:K…a Klebsiella;S…a Salmonella;Uncl…d、U…d Unclassified,未分类的;V…e Vibrionaceae;Ps…es Pseudomonadales;X…s Xanthomonadales;C…m Clostridium;Clo…eae Clostridiaceae;FLavo…aceae Flavobacteriaceae;Flavo…iales Flavobacteriales;Cam…ota Campylobacterota;Camp…ales Campylobacterales;Ba…ta Bacillota;Acti…etes Actinomycetales;Cyan…ceae Cyanophyceae。 Fig. 2 Visualization results of Krona bacteria species annotation for metagenomic sequences of Laelaps jettmari collected from Fengcheng City, Dandong, Liaoning Province |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}