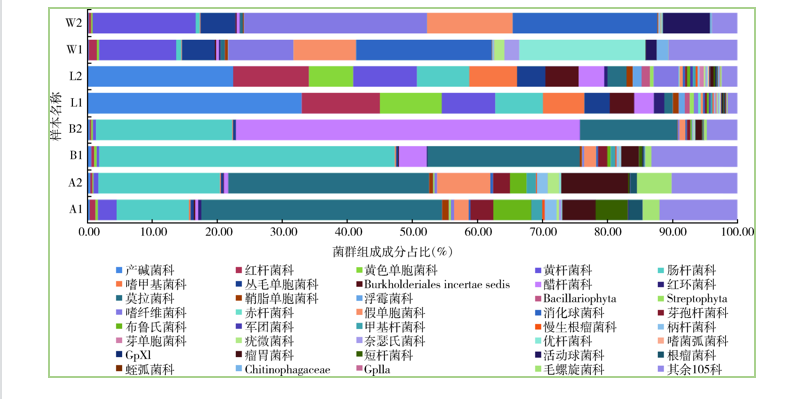
Objective To investigate the differences in the microbial composition in Anopheles sinensis and water in larvae breeding site. Methods An. sinensis adults were collected from Jiading district, Shanghai, China and divided into unfed (A) and fed (B) groups. Water samples were collected from paddy fields in the same area (W). Metagenomic DNA was extracted and the 16S rDNA V4 region was subjected to Illumina Miseq high-throughput sequencing. The sequence number and operational taxonomic units (OTUs) were used for statistical analysis of the composition and abundance of microflorae, the richness and diversity of bacterial communities, and changes in bacterial composition across communities, which were compared with the microbial composition in An. sinensis larvae (L). Results The numbers of high-quality sequences and clustered OTUs were 129 056/2 622.5 (A), 171 734/3 324.5 (B), and 225 890.5/2 997 (W), respectively. The clustered OTUs were assigned to 20 phyla, 144 families, and 295 genera. The bacterial richness as indicated by Chao1 index was in the order of B>W>A. The bacterial diversity was B>W>A (Simpson index) or A>B>W (Shannon index). The dominant bacteria in fed and unfed adults were Proteobacteria and Firmicutes, and the abundant genera were Acinetobacter (A, 41.35%; B, 25.71%) and Pantoea (A, 10.85%; B, 12.70%). Asaia was the most abundant (40.85%) in fed adults. In paddy field water, the dominant bacteria were Firmicutes, Bacteroides, and Proteobacteria. The genera with >10.00% abundances were Desulfosporosinus (24.19%), Flectobacillus (19.48%), Flavobacterium (15.27%), Pseudomonas (12.19%), and Alkalibacter (10.28%). As shown by the t test, the numbers of phyla with statistical differences between A, B, and L were 1 (B/A), 9 (L/A), and 9 (W/A). Principal component analysis showed similar bacterial diversity among samples in the same group, and heatmap showed that W and L clustered into one clade, while B and A into another clade. Conclusion An. sinensis adults are dominated by Acinetobacter, Pantoea, and Asaia. The composition of bacteria in paddy field water is similar to those in larvae living in the water as well as fed and unfed female mosquitos.
[1] 中国科学院中国动物志编辑委员会. 中国动物志.第八卷.昆虫纲. 双翅目:蚊科(上)[M]. 北京:科学出版社,1997:4-33.Editorial Board of Zoology of China,Chinese Academy of Sciences. Fauna sinica.Vol. 8. Insecta. Diptera:Culicidae 1[M]. Beijing:Science Press,1997:4-33.
[2] 张丽,丰俊,夏志贵,等. 2019年全国疟疾疫情特征分析及消除工作进展[J]. 中国寄生虫学与寄生虫病杂志,2020,38(2):133-138. DOI:10.12140/j.issn.1000-7423.2020.02.001.Zhang L,Feng J,Xia ZG,et al. Epidemiological characteristics of malaria and progress on its elimination in China in 2019[J]. Chin J Parasitol Parasit Dis,2020,38(2):133-138. DOI:10.12140/j.issn.1000-7423.2020.02.001.
[3] 曹俊,刘耀宝,曹园园,等. 中国消除疟疾的持续挑战:输入性疟疾[J]. 中国寄生虫学与寄生虫病杂志,2018,36(2):93-96.Cao J,Liu YB,Cao YY,et al. Sustained challenge to malaria elimination in China:imported malaria[J]. Chin J Parasitol Parasit Dis,2018,36(2):93-96.
[4] Gao H,Cui CL,Wang LL,et al. Mosquito microbiota and implications for disease control[J]. Trends Parasitol,2020,36(2):98-111. DOI:10.1016/j.pt.2019.12.001.
[5] Romoli O,Gendrin M. The tripartite interactions between the mosquito,its microbiota and Plasmodium[J]. Parasit Vectors,2018,11:200. DOI:10.1186/s13071-018-2784-x.
[6] Sharma P,Rani J,Chauhan C,et al. Altered gut microbiota and immunity defines Plasmodium vivax survival in Anopheles stephensi[J]. Front Immunol,2020,11:609. DOI:10.3389/fimmu.2020.00609.
[7] Boissière A,Tchioffo MT,Bachar D,et al. Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection[J]. PLoS Pathog,2012,8(5):e1002742. DOI:10.1371/journal.ppat.1002742.
[8] Dong YM,Manfredini F,Dimopoulos G. Implication of the mosquito midgut microbiota in the defense against malaria parasites[J]. PLoS Pathog,2009,5(5):e1000423. DOI:10.1371/journal.ppat.1000423.
[9] Bando H,Okado K,Guelbeogo WM,et al. Intra-specific diversity of Serratia marcescens in Anopheles mosquito midgut defines Plasmodium transmission capacity[J]. Sci Rep,2013,3:1641. DOI:10.1038/srep01641.
[10] Gonzalez-Ceron L,Santillan F,Rodriguez MH,et al. Bacteria in midguts of field-collected Anopheles albimanus block Plasmodium vivax sporogonic development[J]. J Med Entomol,2003,40(3):371-374. DOI:10.1603/0022-2585-40.3.371.
[11] Wang SB,Ghosh AK,Bongio N,et al. Fighting malaria with engineered symbiotic bacteria from vector mosquitoes[J]. Proc Natl Acad Sci USA,2012,109(31):12734-12739. DOI:10.1073/pnas.1204158109/-/DCSupplemental.
[12] Kajla MK. Symbiotic bacteria as potential agents for mosquito control[J]. Trends Parasitol,2020,36(1):4-7. DOI:10.1016/j.pt.2019.07.003.
[13] 李美,汤林华. 中华按蚊实验室种群中肠内细菌菌群分析[J]. 中国寄生虫学与寄生虫病杂志,2010,28(2):143-147.Li M,Tang LH. The midgut bacterial flora in lab-reared Anopheles sinensis[J]. Chin J Parasitol Parasit Dis,2010,28(2):143-147.
[14] 王丹丹. 中华按蚊肠道细菌分子多态性研究[D]. 武汉:华中农业大学,2008.Wang DD. Molecular diversity of the intestine bacterial community of Anopheles sinensis (Diptera:Culicidae)[D]. Wuhan:Huazhong Agricultural University,2008.
[15] 翟小战. 中华按蚊肠道细菌群落的研究[D]. 武汉:华中农业大学,2014.Zhai XZ. The intestinal bacterial flora community structure of Anopheles sinensis mosquitoes (Diptera:Culicidae)[D]. Wuhan:Huazhong Agricultural University,2014.
[16] 南春燕,马雅军,徐建农,等. 中华按蚊幼虫肠道细菌宏基因组的组成研究[J]. 中国寄生虫学与寄生虫病杂志,2013,31(2):114-119.Nan CY,Ma YJ,Xu JN,et al. Taxonomic composition of metagenomic community in the larval gut of mosquito Anopheles sinensis (Diptera:Culicidae)[J]. Chin J Parasitol Parasit Dis,2013,31(2):114-119.
[17] Gao H,Bai L,Jiang YM,et al. A natural symbiotic bacterium drives mosquito refractoriness to Plasmodium infection via secretion of an antimalarial lipase[J]. Nat Microbiol,2021,6(6):806-817. DOI:10.1038/s41564-021-00899-8.
[18] Bai L,Wang LL,Vega-Rodríguez J,et al. A gut symbiotic bacterium Serratia marcescens renders mosquito resistance to Plasmodium infection through activation of mosquito immune responses[J]. Front Microbiol,2019,10:1580. DOI:10.3389/fmicb.2019.01580.
[19] Tran Q,Pham DT,Phan V. Using 16S rRNA gene as marker to detect unknown bacteria in microbial communities[J]. BMC Bioinformatics,2017,18(14):499. DOI:10.1186/s12859-017-1901-8.
[20] Wang Y,Gilbreath Ⅲ TM,Kukutla P,et al. Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya[J]. PLoS One,2011,6(9):e24767. DOI:10.1371/journal.pone.0024767.
[21] Champion CJ,Xu JN. The impact of metagenomic interplay on the mosquito redox homeostasis[J]. Free Radic Biol Med,2017,105:79-85. DOI:10.1016/j.freeradbiomed.2016.11.031.
[22] Gimonneau G,Tchioffo MT,Abate L,et al. Composition of Anopheles coluzzii and An. gambiae microbiota from larval to adult stages[J]. Infect Genet Evol,2014,28:715-724. DOI:10.1016/j.meegid.2014.09.029.
[23] Osei-Poku J,Mbogo CM,Palmer WJ,et al. Deep sequencing reveals extensive variation in the gut microbiota of wild mosquitoes from Kenya[J]. Mol Ecol,2012,21(20):5138-5150. DOI:10.1111/j.1365-294X.2012.05759.x.
[24] 彭恒,陈翰明,陈辉莹,等. 我国赫坎按蚊种团的分子鉴别及中华按蚊的区系分布研究[J]. 中国寄生虫学与寄生虫病杂志,2020,38(1):58-66,73. DOI:10.12140/j.issn.1000-7423.2020.01.009.Peng H,Chen HM,Chen HY,et al. Molecular identification of Anopheles hyrcanus group and faunal distribution of An. sinensis (Diptera:Culicidae) in China[J]. Chin J Parasitol Parasit Dis,2020,38(1):58-66,73. DOI:10.12140/j.issn.1000-7423.2020.01.009.
[25] Wang Y,Qian PY. Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies[J]. PLoS One,2009,4(10):e7401. DOI:10.1371/journal.pone.0007401.
[26] Li KL,Chen HY,Jiang JJ,et al. Diversity of bacteriome associated with Phlebotomus chinensis (Diptera:Psychodidae) sand flies in two wild populations from China[J]. Sci Rep,2016,6:36406. DOI:10.1038/srep36406.
[27] de O Gaio A,Gusmão DS,Santos AV,et al. Contribution of midgut bacteria to blood digestion and egg production in Aedes aegypti (Diptera:Culicidae) (L.)[J]. Parasit Vectors,2011,4:105. DOI:10.1186/1756-3305-4-105.
[28] Zouache K,Raharimalala FN,Raquin V,et al. Bacterial diversity of field-caught mosquitoes,Aedes albopictus and Ae. aegypti,from different geographic regions of Madagascar[J]. FEMS Microbiol Ecol,2011,75(3):377-389. DOI:10.1111/j.1574-6941.2010.01012.x.
[29] Alfano N,Tagliapietra V,Rosso F,et al. Changes in microbiota across developmental stages of Aedes koreicus,an invasive mosquito vector in Europe:indications for microbiota-based control strategies[J]. Front Microbiol,2019,10:2832. DOI:10.3389/fmicb.2019.02832.
[30] Akorli J,Namaali PA,Ametsi GW,et al. Generational conservation of composition and diversity of field-acquired midgut microbiota in Anopheles gambiae (sensu lato) during colonization in the laboratory[J]. Parasit Vectors,2019,12:27. DOI:10.1186/s13071-019-3287-0.
[31] Kalappa DM,Subramani PA,Basavanna SK,et al. Influence of midgut microbiota in Anopheles stephensi on Plasmodium berghei infections[J]. Malar J,2018,17:385. DOI:10.1186/s12936-018-2535-7.
[32] Zoure AA,Sare AR,Yameogo F,et al. Bacterial communities associated with the midgut microbiota of wild Anopheles gambiae complex in Burkina Faso[J]. Mol Biol Rep,2020,47:211-224. DOI:10.1007/s11033-019-05121-x.
[33] Coon KL,Vogel KJ,Brown MR,et al. Mosquitoes rely on their gut microbiota for development[J]. Mol Ecol,2014,23(11):2727-2739. DOI:10.1111/mec.12771.
[34] Feng XY,Huang LB,Lin L,et al. Genetic diversity and population structure of the primary malaria vector Anopheles sinensis (Diptera:Culicidae) in China inferred by cox1 gene[J]. Parasit Vectors,2017,10:75. DOI:10.1186/s13071-017-2013-z.
[35] Gabrieli P,Caccia S,Varotto-Boccazzi I,et al. Mosquito trilogy:microbiota,immunity and pathogens,and their implications for the control of disease transmission[J]. Front Microbiol,2021,12:630438. DOI:10.3389/fmicb.2021.630438.
[36] Mancini MV,Damiani C,Accoti A,et al. Estimating bacteria diversity in different organs of nine species of mosquito by next generation sequencing[J]. BMC Microbiol,2018,18:126. DOI:10.1186/s12866-018-1266-9.

